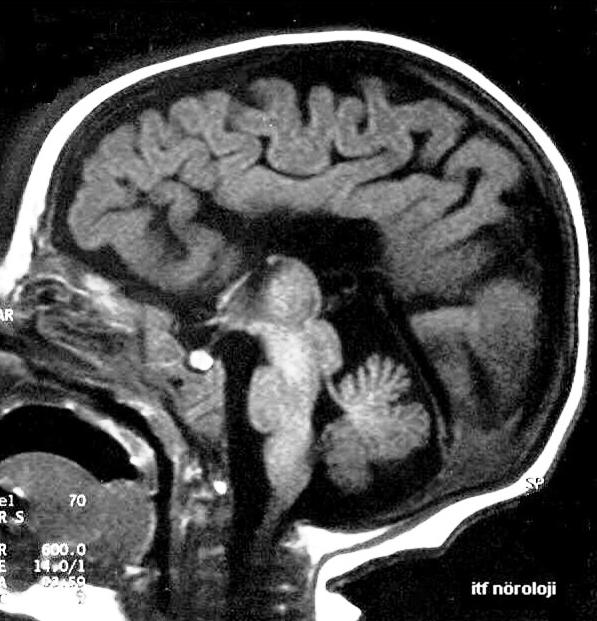
Korpus kallozum (Corpus callosum) beynin iki yarısını (iki hemisferi) bağlayan bir yapının gelişmemesine denir.
1 ) Korpus Kallozum Agenezisi Nedir?
Korpus Kallozum Agenezisi (ACC), çok az rastlanan bir doğumsal anomalidir.
Korpus kallozum, bir kısmı veya tamamamı gelişme sağlamamıştır. Korpus kallozum, beynin iki hemisferini bağlamaya yarayan temel yoldur. Korpus kallozumun gelişimi, anne karnında ki bebeğin, yaşamın 5. haftasında primitif lamina terminalisin oluşumu ile beraber başlar.
Primitif lamina terminalis kalınlaşıp, komissural plağını meydana getirir. Glial hücreler birleşerek köprüye benzeyen bir yapıyı ortaya çıkarır. Bu yapı, longitudinal fissürü geçip beynin karşı yarısında bulunan yerine giden kallozal liflere yol gösterir. Korpus kallozun hamilelik döneminin 17. Haftası içerisinde matür hale gelir. Korpus kallozum agenezisinde (KKA) komissural lifler orta hattı geçemez.
Korpus kallozum yerine lateral ventriküllerin medialinde Probst band olarak ifade edilen ismi ile arkaya doğru ilerleyen kör bir band ortaya çıkarır. Probst band, hemisferler içinde longitudinal bağlantının gerçekleşmesine yardımcı olan liflerdir. Bu bandlar, lateral ventriküllerin ön boynuzlarını ayırmaya yarar.
2 ) Diğer Kallozal Anomaliler nelerdir?
Korpus kallozum agenezisi haricinde, diğer kallozal anomalilerin içerisinde;
Korpus kallozumun hipogenezi(parsiyel formasyon), disgenezi (malformasyon) ve hipoplazisi sayılabilir.
3 ) Tanı Nasıl Yapılır?
Kallozal hastalıklar, sadece beyin taraması ile tanınabilir. MR, BT, prenatal ultrason ve prenatal MR ile de tanı gerçekleştirilebilir.
4 ) Korpus Kallozum Agenezisi oluşma nedeni nedir?
KKA, izole bir bulgu olabilir. Ancak daha çok başka malformasyonlar, kromozomal aberasyon ve metabolizma bozukluklarını içeren genetik sendromlar ile ilişkili olarak görülebilir.
5 ) İlişkili anomalileri Nelerdir?
Santral sinir sistemi anomalileri
Chiari malformasyonları
Sinir migrasyon anomalileri
Lizensefali
Şizensefali
Pakigiri
Polimikrogiri
Ensefalosel
Dandy-Walker malformasyonu
Holoprozensefali
Olivopontoserebellar dejenerasyon
Yüz anomalileri
Kardiyovasküler anomaliler
Genitoüriner anomaliler
Gastrointestinal anomaliler
Respiratuvar anomaliler
Muskuloskeletal anomaliler
Korpus Kallozum Agenezisinde fetüse yapılacak herhangi bir müdahale yoktur.
6 )Fetüse Müdahale Nasıl Yapılır?
Korpus Kallozum Agenezisinde, şuan için fetüse herhangi bir müdahalenin yapılması söz konusu değildir.
7 ) Yeni doğan bebeğin tedavisi nasıl gerçekleştirilir?
Yeni doğan bebeğin, ayrıntılı bir şekilde muayene edilmesi gereklidir. Yeni doğana MR yapılması da, tedavinin tanısı için gayet uygundur. MR ile beraber, eşlik eden diğer SSS anomalileri ayrıntılı bir şekilde teşhis edilebilir. Bu konuda MR, BT ve USG den çok daha başarılıdır. Yeni doğan bebekte, metabolik hastalıklardan şüphelenilmesi halinde, araştırma yapılması gerekmektedir.
8 ) Cerrahi Tedavi Nasıl Uygulanır?
İzole KKA için cerrahi tedavi söz konusu değildir. KKA beraberinde gelen intrakranial ve ekstrakranial anomaliler için, cerrahi müdahaleye ihtiyaç duyulup duyulmadığı değerlendirme altına alınır. Progresif ventriküler dilatasyon ve artmış kafa büyümesi halinde, ventriküloperitoneal şanta ihtiyaç duyulabilir.
9 ) Tekrar etme ve genetik
KKA’nin tekrar nüks etme riski, metabolik hastalık veya genetik sendroma eşlik etmesi veya izole olması ile alakalıdır. Eğer KKA, anöploidi ile alakalı ise, yeniden tekrarlama riski %1 veya annenin yaşı ile alakalı olur. Hangisi daha fazla ise, gerçek tekrarlama riski odur. KKA izole ise, tekrarlama riski yaklaşık olarak %2 ile %3 civarındadır.
KKA, çeşitli sendromların tanısı için bilinen bir kriterdir.
Aicardi Sendromu
Andermann Sendromu
Akrokallozal Sendrom
KKA daha çok sporodik olarak bilinse de, ailesel vakalar da yayınlanmıştır.
10 ) Hamileliğin İdaresi Nasıl Sağlanmalıdır?
KKA tespit edilen olgularda, intrakranial ve ekstrakranial anomaliler dikkatli bir şekilde incelenmelidir. Olguların yaklaşık %70’inde 18, 8 ve 13 trizomi çok fazla görüldüğünden dolayı ve 8 trizominin tek bulgusu KKA olduğu için, ek anomali bulunmasa bile, bütün olgulara amniyosentez yapılması gerekmektedir.
KKA izole bir bulgu ve kromozom anomalisi bulunmuyor ise, standart obstetrik bakımı değiştirmeye de ihtiyaç duyulmaz. Bu tanı, üçüncü basamakta doğumun gerçekleştirilmesini gerektiren bir anomali değildir. Doğumun tamamlanmasının ardından, genetik ve pediatrik nöroloji uzmanı tarafından konsülte edilmesi gerekmektedir. Beraberinde gelen başka anomalilerin bulunması halinde, üçüncü basamakta doğum sağlanmalı ve anomalinin görüldüğü ilgili branşta pediatrist tarafından konsülte edilmelidir.
11 ) Doğum nasıl olmalı?
Hidrosefali veya makrosefali bulunmuyor ise, vajinal doğumun yapılması tavsiye edilmektedir. Obstetrik endikasyon yok ise, erken doğum yaptırılmasına ihtiyaç duyulmaz.




Deprecated: Function get_magic_quotes_gpc() is deprecated in /home/coolkadin/public_html/wp-content/themes/tomasdaisy/framework/lib/eltd.functions.inc on line 284
Deprecated: Function get_magic_quotes_gpc() is deprecated in /home/coolkadin/public_html/wp-content/themes/tomasdaisy/framework/lib/eltd.functions.inc on line 284
Deprecated: Function get_magic_quotes_gpc() is deprecated in /home/coolkadin/public_html/wp-content/themes/tomasdaisy/framework/lib/eltd.functions.inc on line 284